To obtain permission to market a drug, the manufacturer must satisfy the FDA that the drug is both safe and effective. Additional testing often enhances safety and effectiveness, but requiring a lot of testing has at least two negative effects. First, it delays the arrival of superior drugs. During the delay, some people who would have lived end up dying. Second, additional testing requirements raise the costs of bringing a new drug to market; hence, many drugs that would have been developed are not, and all the people who would have been helped, even saved, are not.
In addition, because FDA approval is mandatory, industry and medicine must heed FDA standards regardless of their relevance, efficiency, and appropriateness. Not all testing is equally beneficial. The FDA apparatus mandates testing that, in some cases, is not useful or not appropriately designed. The case against the FDA is not that premarket testing is unnecessary but that the costs and benefits of premarket testing would be better evaluated and the trade-offs better navigated in a voluntary, competitive system of drug development.
Three bodies of evidence indicate that the costs of FDA requirements exceed the benefits. In other words, three bodies of evidence suggest that the FDA kills and harms, on net. First, we compare pre-1962 drug approval times and rates of drug introduction with post-1962 approval times and rates of introduction. Second, we compare drug availability and safety in the United States with the same in other countries. Third, we compare the relatively unregulated market of off-label drug uses in the United States with the on-label market. In the final section, before turning to reform options, we also discuss the evidence showing that the costs of FDA advertising restrictions exceed the benefits.
Comparison of Pre- and Post-1962
Sam Peltzman (1973) wrote the first serious cost-benefit study of the FDA. He focused his attention on the 1962 Kefauver-Harris Amendments to the Food, Drug, and Cosmetics Act of 1938, which significantly enhanced FDA powers. The amendments added a proof-of-efficacy requirement to the existing proof-of-safety requirement, removed time constraints on the FDA disposition of NDAs, and gave the FDA extensive powers over the clinical testing procedures drug companies used to support their applications.
Using data from 1948 to 1962, Peltzman created a statistical model to predict the yearly number of new drug introductions. The model is based on three variables, the most important of which is the size of the prescription drug market, lagged two years. The idea is that if the prescription drug market were large two years ago, manufacturers would invest more money in research and development, which would pay off two years later in a new drug. (Prior to 1962, it took approximately two years to develop a new drug.) Despite the model ’s simplicity, it tracks the actual number of new drug introductions quite well, as indicated by figure 2.
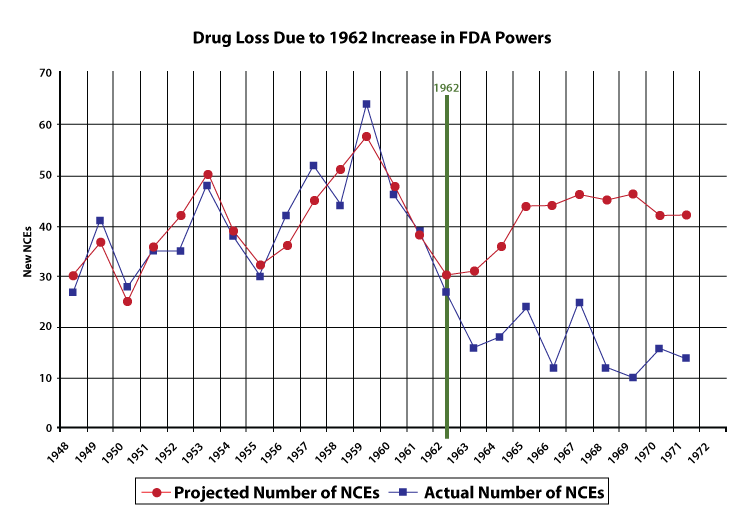
Because Peltzman’s model tracks the pre-1962 drug market quite well, we have some confidence that if all else had remained equal, the model also should have roughly tracked the post-1962 drug market. Peltzman’s model, in other words, estimates the number of new drugs that would have been produced if the FDA’s powers had not been increased in 1962. Thus, by comparing the model results with the actual number of new drugs, we can draw an estimate of the effect of the 1962 amendments. The model predicts a probable post-1962 average of forty-one new chemical entities (NCEs, or new drugs) approved per year.
The average number of new drugs introduced pre-1962 (forty) was also much larger than the post-1962 average (sixteen). Thus, whether one compares pre- and post-1962 averages or compares the results from a forecast with the actual results, the conclusions are the same: the 1962 Amendments caused a significant drop in the introduction of new drugs. Using data of longer span, Wiggins (1981) also found that increased FDA regulations raised costs and reduced the number of new drugs.
Even if FDA regulations have not improved safety, they might be redeemed if they have reduced the proportion of inefficacious drugs on the market. Using a variety of tests, however, Peltzman (1973) found little evidence to suggest a decline in the proportion of inefficacious drugs reaching the market since 1962. Thus, he concluded, “(the) penalties imposed by the marketplace on sellers of ineffective drugs prior to 1962 seem to have been enough of a deterrent to have left little room for improvement by a regulatory agency” (1086). Similarly, in their survey of the literature, Grabowski and Vernon (1983) conclude, “In sum, the hypothesis that the observed decline in new product introductions has largely been concentrated in marginal or ineffective drugs is not generally supported by empirical analyses” (34).
The costs of FDA regulations do not vary with the number of potential users of the drug, so the decline in drug development has been especially important in the treatment of rare diseases. By definition, each rare disease afflicts only a small number of people, but there are thousands of rare diseases. In aggregate, rare diseases afflict millions of Americans: according to an AMA estimate (AMA 1995), as many as 10 percent of the population. Thus, millions of Americans have few or no therapies available to treat their diseases because of increased costs of drug development brought about by stringent FDA “safety and efficacy” requirements. In response to this problem, in 1983 the Orphan Drug Act was passed to provide tax relief and exclusive privileges to firms developing drugs for diseases affecting two hundred thousand or fewer Americans (AMA 1995). It would be better to reduce or eliminate FDA regulations for all drugs and patient populations.
The Grisly Comparison
The delay and large reduction in the total number of new drugs has had terrible consequences. It is difficult to estimate how many lives the post-1962 FDA controls have cost, but the number is likely to be substantial; Gieringer (1985) estimates the loss of life from delay alone to be in the hundreds of thousands (not to mention millions of patients who endured unnecessary morbidity). When we look back to the pre-1962 period, do we find anything like this tragedy? The historical record—decades of a relatively free market up to 1962—shows that voluntary institutions, the tort system, and the pre-1962 FDA succeeded in keeping unsafe drugs to a low level. The Elixir Sulfanilamide tragedy, in which 107 people died, was the worst of those decades. Every life lost is important, but the grisly comparison is necessary. The number of victims of Elixir Sulfanilamide tragedy and of all other drug tragedies prior to 1962 is very small compared to the death toll of the post-1962 FDA.
Comparison with Other Countries
The second source of evidence comes from comparing drug availability and safety in the United States with the same in other countries. Prior to the Kefauver-Harris Bill of 1962, the average time from the filing of an IND to approval was seven months. By 1967, however, the average time to approval had increased to thirty months. Time to approval continued to rise through the late 1970s, when on average a successful drug took more than ten years to get approved. In the late 1980s and 1990s, times to approval decreased somewhat, but are still eight years on average, far higher than in the 1960s (Peltzman 1974; Thomas 1990; Tufts Center for the Study of Drug Development 1998).
Time to approval has historically been shorter by years in Europe than in the United States. Drugs are usually available in Europe before they are available in the United States. The difference between the time of a drug’s availability in Europe and that in the United States has come to be called the drug lag (Grabowski 1980; Kaitin et al. 1989; Wardell 1973, 1978a,1978b; Wardell and Lasagna 1975). In recent years, however, the FDA has improved. In the latest data, covering 1996–98, the average time from filing the IND to submitting the NDA was 5.9 years and average NDA approval time was 1.4 years, for a total of 7.3 years, the quickest approval times in decades (Kaitin and Healy 2000). Researchers have suggested that the drug lag may be disappearing (Healy and Kaitin 1999).
What is significant for our purposes is that from approximately 1970 to 1993 the FDA clearly lagged significantly behind its counterparts in the United Kingdom, France, Spain, and Germany (Kaitin and Brown 1995). This fact gives us a basis for comparison: During the period of consistent drug lag, did the delay correspond to greater safety? Put another way: Did speedier drug approval in Europe lead to a scourge of unsafe drugs?
If the U.S. system resulted in appreciably safer drugs, we would expect to see far fewer postmarket safety withdrawals in the United States than in other countries. Bakke et al. (1995) compared safety withdrawals in the United States with those in Great Britain and Spain, each of which approved more drugs than the United States during the same time period. Yet, approximately 3 percent of all drug approvals were withdrawn for safety reasons in the United States, approximately 3 percent in Spain, and approximately 4 percent in Great Britain. There is no evidence that the U.S. drug lag brings greater safety. Wardell and Lasagna (1975) concluded their comparison of drug approvals in the United States and Great Britain by noting: “In view of the clear benefits demonstratable [sic] from some of the drugs introduced into Britain, it appears that the United States has lost more than it has gained from adopting a more conservative approach” (105).
Deaths owing to drug lag have been numbered in the hundreds of thousands. Wardell (1978a) estimated that practolol, a drug in the beta-blocking family, could save ten thousand lives a year if allowed in the United States. Although the FDA allowed a first beta-blocker, propranolol, in 1968, three years after that drug had been available in Europe, it waited until 1978 to allow the use of propranolol for the treatment of hypertension and angina pectoris, its most important indications. Despite clinical evidence as early as 1974, only in 1981 did the FDA allow a second beta-blocker, timolol, for prevention of a second heart attack. The agency’s withholding of beta-blockers was alone responsible for probably tens of thousands of deaths (on this general issue see Gieringer 1985; Kazman 1990).
A chief source of information about drug delay is the Tufts Center for the Study of Drug Development, a scholarly, not too outspoken research center funded chiefly by pharmaceutical companies. Their information is often mined by researchers at the Competitive Enterprise Institute (CEI). The CEI has noted that in recent years thousands of patients have died because the FDA has delayed the arrival of new drugs and devices, including interleukin-2, taxotere, vasoseal, ancrod, glucophage, navelbine, lamictal, ethyol, photofrin, rilutek, citicoline, panorex, femara, prostar, omnicath, and transform. Prior to FDA approval, most of these drugs and devices had already been available in other countries for a year or longer.
Gieringer (1985) used data on drug disasters in countries with less-stringent drug regulations than the United States to create a ballpark estimate of the number of lives saved by the extra scrutiny induced by FDA requirements. He then computed a similar ballpark figure for the number of lives lost owing to drug delay:
[T]he benefits of FDA regulation relative to that in foreign countries could reasonably be put at some 5,000 casualties per decade or 10,000 per decade for worst-case scenarios. In comparison, it has been argued above that the cost of FDA delay can be estimated at anywhere from 21,000 to 120,000 lives per decade. . . . Given the uncertainties of the data, these results must be interpreted with caution, although it seems clear that the costs of regulation are substantial when compared to benefits. (196)
Note three things about the foregoing passage. (1) The comparison is between the FDA and the foreign systems of drug control. (2) The relative benefits of the FDA are expressed in number of casualties, whereas the relative costs are in number of lives. (3) In addressing the costs, Gieringer estimated the costs only from drug delay; he does not attempt to quantify the costs associated with drug loss. Nevertheless, his conclusion is clear: the FDA is responsible for more lives lost than lives saved.
FDA Incentives
Even if the FDA required only the most relevant clinical trials and worked at peak efficiency to evaluate new drugs, the trade-off between more testing and delayed drugs would still exist. We cannot escape this trade-off. The only question is whether a centralized bureaucracy should decide on these trade-offs for everyone or patients and doctors should make such decisions. Much of the FDA’s delay, however, is not owing to useful—albeit not necessarily optimal—testing. Much of the delay is pure waste. The cause is not laziness but incentives. (Read the sidebar Why the FDA Has an Incentive to Delay the Introduction of New Drugs for an explanation.)
Comparison of On-Label and Off-Label Usage
The third sort of evidence on the costs of FDA regulations comes from comparing the utilization of drugs for their on-label uses with their utilization for off-label uses. The hidden lesson of off-label usage is that, even in today’s highly regimented setting, there functions a realm of efficacy testing and assurance quite apart from the FDA.
When the FDA evaluates the safety and efficacy of a drug, the evaluation is made with respect to a specified use of the drug. Once a drug has been approved for some use, however, doctors may legally prescribe the drug for other uses. Approved uses are known as on-label uses, and other uses are considered off-label uses. Amoxicillin, for example, has an on-label use for treating respiratory tract infections and an off-label use for treating stomach ulcers.
For the on-label treatment of respiratory tract infections, amoxicillin has been tested and passed muster in all three phases of the IND clinical study; phase I trials for basic safety and phase II and phase III trials for efficacy. For the treatment of stomach ulcers, however, amoxicillin has not gone through FDA-mandated phase II and phase III trials and thus is not FDA approved for this use. Indeed, amoxicillin will probably never go through FDA efficacy trials for the treatment of stomach ulcers because the basic formulation is no longer under patent. Yet any textbook or medical guide discussing stomach ulcers will mention amoxicillin as a potential treatment, and a doctor who did not consider prescribing amoxicillin or other antibiotic for the treatment of stomach ulcers would today be considered highly negligent. Off-label uses are in effect regulated according to the FDA’s pre-1962 rules (which required only safety, not efficacy), whereas on-label uses are regulated according to the post-1962 rules.
FDA defenders suggest that an unregulated market for drugs would be a medical disaster. Do patients and doctors shrink in fear from uses not certified by the FDA?
Not at all! Most hospital patients are given drugs that are not FDA approved for prescribed use. In a large number of fields, a majority of patients are prescribed at least one drug off-label. Off-label prescriptions are especially common for AIDS, cancer, and pediatric patients, but are also common throughout medicine.
Doctors learn of off-label uses from extensive medical research, testing, peer-reviewed publications, newsletters, lecture presentations, conferences, advertising, Internet sources, and trusted colleagues. Scientists and doctors, working through professional associations and organizations, make official determinations of “best practice” and certify off-label uses in standard reference compendia such as AMA Drug Evaluations, American Hospital Formulary Service Drug Information, and U.S. Pharmacopoeia Drug Indications. Doctors use this information to try to make the best decisions for their patients. Medical decisions are most often made under uncertainty and partial ignorance, so there is rarely a single best decision, and different doctors and different patients choose different treatments. New information constantly flows into this system as outcomes accumulate, epidemiological studies reveal new correlations, scientists propose theoretical explanations, researchers design and embark on new clinical studies, scientific institutions arrive at new judgments, and pharmaceutical companies create new drugs. As this medical knowledge grows and develops, information flows in a decentralized fashion, and doctors adjust their decisions accordingly. (See the section The Sensible Alternative on how the institutions work without government intervention.)
Economist J. Howard Beales (1996) found that off-label uses appeared in the Pharmacopoeia on average 2.5 years earlier than the FDA recognized those uses. The difference between the on-label and off-label markets is not that the off-label market is “unregulated" but that it is unregulated by the FDA, a centralized and coercive authority. In approving or rejecting a new drug, the FDA makes a decision everyone must obey. It’s as if the Department of Transport unilaterally decided what vehicles Americans could and could not purchase. Heterogeneity among patients in both preferences and circumstances is great. A drug that can save the life of A may be dangerous to B even if A and B have the same disease. An athlete and a college professor with the same disease may choose different courses of treatment. The FDA’s “one size fits all” policy is not appropriate for every patient.
The off-label market is regulated by thousands of doctors and patients acting in a decentralized manner. Compared to the FDA, this market adjusts quickly to new information, shows less sign of biased incentives, and allows a more precise adjusting of treatment decisions to preferences and the conditions of time and place. The evidence indicates that these benefits are not offset by significantly greater risk (Tabarrok 2000). The off-label market operates with much less government intervention than the on-label market and provides a good idea of the benefits to be had from reducing FDA control over approval decisions.
By their actions, doctors tell us that they believe in off-label prescribing. Getting the FDA to approve a new use for an old drug requires an expensive and lengthy process. In many cases, the costs to the sponsor of the required testing exceed the benefits of approval. It is clear that if the FDA prohibited off-label prescribing, current practices would have to change significantly. No one would be foolish enough to suggest that the FDA prohibit off-label prescribing.
But there is a logical inconsistency in allowing off-label prescribing and requiring proof of efficacy for the drug’s initial use (Tabarrok 2000). Logical consistency would require us either (1) to oppose off-label prescribing and favor initial proof of efficacy, or (2) to favor off-label prescribing and oppose initial proof of efficacy. Experience recommends the second option. Efficacy requirements should be dropped altogether!
Summary of the Three Bodies of Evidence: FDA-Caused Mortality and Morbidity Are Unredeemed
Evidence from the pre-1962 market shows that FDA restrictions have greatly reduced the number of new drugs, and because there was little or no corresponding gain in drug quality, the concomitant mortality and morbidity were unredeemed. The international evidence shows that there has long been a drug lag in the United States, and because Americans have not benefited from the extra “precaution,” the concomitant mortality and morbidity are unredeemed. Finally, the off-label evidence indicates that the network of doctors, patients, pharmaceutical firms, hospitals, universities, rating organizations, and so forth is really in charge of defining and judging efficacy and that it functions smoothly and successfully in the realm of uses not approved by the FDA; hence, the mortality and morbidity that result from proof-of-efficacy requirements are unredeemed. All the systematic evidence goes against the coercive FDA apparatus.
FDA Advertising Restrictions: Ignorance Is Death
In addition to permitting drugs on the market, the FDA controls advertising and promotion. The costs of such control parallel the costs of restricting drugs. They include (1) reducing the speed at which consumers learn of and adopt important new therapies; (2) reducing the size of the market for drugs, thereby reducing the incentive to research and develop new drugs; and (3) reducing the number of treatment options, making it more difficult for physicians to provide therapies tailored to each individual patient (Rubin 1995; Tabarrok 2000).
In numerous instances, the FDA has reduced the speed at which patients and their agents have learned of and adopted new drugs or new uses of old drugs. The most important example is aspirin.
The FDA prevented aspirin manufacturers from advertising that clinical studies had shown that the use of aspirin during and after heart attacks might prevent death. When, years after the clinical studies had been completed, the FDA finally sanctioned aspirin for heart-attack patients, Dr. Carl Pepine, codirector of cardiovascular medicine at the University of Florida College of Medicine, estimated that as many as ten thousand lives annually could be saved. In other words, Dr. Pepine thought that the FDA restrictions preventing the advertising and promotion of aspirin for heart attack patients were responsible for the deaths of tens of thousands of people. Noting that the decision should have come years earlier, Dr. Pepine said, “I’m disappointed that something that has such potential to save so many lives took so long. But it’s better late than never” (quoted in Ross 1996). Paul Rubin (1995), whose paper on FDA advertising restrictions provides the title for this section, wrote that “the banning of advertising of aspirin for first heart attack prevention, may be the single most harmful regulatory policy currently pursued by any agency of the U.S. government” (48). (Keith [1995] reaches a similar, though less pointed, conclusion.) Despite studies showing benefits, the FDA still does not allow aspirin manufacturers to advertise the benefits of aspirin as a preventive measure for people at high risk for a first heart attack.
Another example: In 1992, the federal Centers for Disease Control and Prevention (CDC) recommended that women of childbearing age take folic acid supplements. Studies showed that taking folic acid reduced risks of babies suffering neural-tube birth defects such as anencephaly and spina bifida. The FDA immediately announced, however, that it would prosecute any food or vitamin manufacturer that placed the CDC recommendation in its advertising or product labeling (Calfee 1997). The public did not learn of the importance of folic acid until Congress passed the Dietary Supplement Health and Education Act of 1994, which loosened the FDA’s vise on the advertising of vitamins and other dietary supplements. Within only a few years of its ban on publicizing the CDC recommendation, the FDA made a complete turnabout. Since 1998, the agency has required manufacturers to fortify a variety of grain products with folic acid—that which is not prohibited is mandatory!
The FDA has also restricted how manufacturers can promote the off-label uses of drugs (these restrictions were in part ruled unconstitutional in Washington Legal Foundation v. Friedman). Such restrictions make it more difficult for doctors to best match patient with treatment. In a survey, 79 percent of neurologists and neurosurgeons, 67 percent of cardiologists, and 76 percent of oncologists said that the FDA should not restrict information about off-label uses. In response to a follow-up question, similar numbers indicated that the FDA policy of limiting information had made it more difficult for them to learn about new uses of drugs and devices (Conko 1998).
(On government control of advertising more generally, see Calfee 1997; Kaplar 1993; Masson and Rubin 1985; Rubin 1991a, 1991b, 1994; and Tabarrok 2000; they evaluate FDA restrictions on advertising and promotion in more detail.)
In recent years, the courts have found First Amendment limits on the FDA’s power to restrict commercial speech. In Washington Legal Foundation v. Friedman, the Federal Appeals Court for the District of Columbia ruled that the FDA may not prohibit drug manufacturers from providing practitioners and others with independent publications such as off-prints of scientific articles, or from organizing medical education programs about off-label uses of their products. The court’s ruling contained strong and decisive language:
“In asserting that any and all scientific claims about the safety, effectiveness, contraindications, side effects, and the like regarding prescription drugs are presumptively untruthful or misleading until the FDA has had the opportunity to evaluate them, FDA exaggerates its overall place in the universe . . . the conclusions reached by a laboratory scientist or university academic and presented in a peer-reviewed journal or textbook, or the findings presented by a physician at a CME seminar are not “untruthful” or “inherently misleading” merely because the FDA has not yet had the opportunity to evaluate the claim. As two commentators astutely stated, ‘the FDA is not a peer review mechanism for the scientific community.’ ”
In Pearson v. Shalala the Federal appeals court for the District of Columbia followed this reasoning and ruled that dietary supplements can be labeled with health claims so long as they bear a disclaimer that such claims have not received FDA approval.